
Designing Medicine, One Binder at a Time
Johannes Weniger, Geeve George · Berlin/Bengaluru
Today we're announcing two things.
The first is a computational pipeline that designs a therapeutic peptide binder for a protein target in under an hour, for under $7.
The second is the autonomous robotic lab that synthesizes those designs, validates them, and feeds the results back into the model, closing the loop between an idea and an experiment.
Together, they're the beginning of something we've wanted to exist for a long time: a system where designing a new medicine is a thing you run, not a thing you wait years to attempt.
And we don't believe the breakthroughs will come from computation alone. In silico design has a ceiling — a model can only ever be as good as the data it has seen, and prediction is not proof. The real gains come from closing the loop: every binder the robot synthesizes and tests becomes hard wet-lab evidence fed back into the model, carrying it somewhere pure simulation never could reach.
This is what we're building at BIOS.
Why this matters
For most of history, finding a drug was guesswork.
You had a hunch about a molecule, you made it, and you tested it in cells and animals, with very little idea, going in, whether it would work. Each loop took years and cost a fortune. The graveyard of failed candidates is enormous, and most of biology has never been drugged at all.
We think that era is ending.
We can now predict the three-dimensional shape of almost any protein. We can generate new molecules designed to fit it. And we can score those designs computationally before a single one touches a lab bench. The part that used to take a building full of scientists and a decade of work is starting to look like a pipeline you can run on a laptop and a cluster.
When you can design candidates this fast and this cheaply, the bottleneck stops being human hands and starts being how fast we can think.
We're learning to write the code of life.
The mental model: locks and keys
Here's the whole field in one analogy.
A receptor on the surface of a cell is a lock. It has many keyholes: places on its surface where another molecule can slot in. When the right molecule fits the right keyhole, the lock turns, and something happens in the body: a signal fires, a hormone releases, hunger fades, blood sugar drops.
A binder is a key. It's a small protein, a peptide, shaped to fit one specific keyhole and turn that one specific lock.
Designing a medicine, in this framing, is two problems:
- Find the keyhole. Which spot on the protein should we aim for?
- Make the key. Build a molecule that fits it, holds on tightly, and doesn't accidentally open every other lock in the body.
Finding the keyhole is maybe 10% of the work; for well-studied proteins, much of it is already known. Making the key is the other 90%, and for most of history it couldn't be done inside a computer at all.
That just changed. A wave of machine-learning breakthroughs cracked the problem open. First AlphaFold solved protein structure prediction, going from amino-acid sequence to 3D fold, well enough to win the 2024 Nobel Prize in Chemistry. Then generative models learned to run it in reverse: RFdiffusion shapes entirely new backbones out of noise, ProteinMPNN picks the sequence that will fold into each one, and a new generation of designers can invent proteins that have never existed in nature. For the first time, designing a binder is a computation you run, not a decade of bench work you have to survive. Harnessing that frontier, orchestrating those models end to end, fully autonomously, is what BIOS does.
Let's walk through it on a real target.
A worked example: GLP-1R
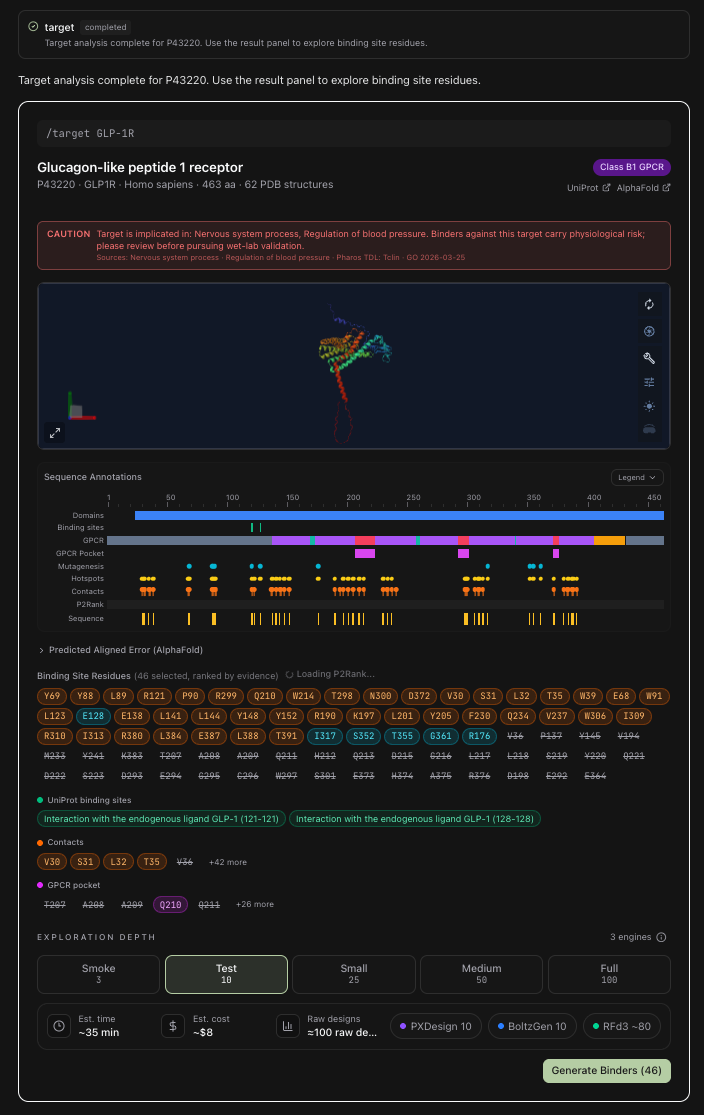
We'll use GLP-1R, the glucagon-like peptide-1 receptor (UniProt P43220, a Class B1 GPCR, 463 amino acids long).
You've met this protein before, even if you don't know it. It's the receptor behind the wave of GLP-1 medicines that regulate appetite, insulin, and blood sugar. Activate it, and your body feels less hungry and manages glucose better. It's one of the most consequential drug targets of the decade, and a perfect example, because it's exceptionally well studied.
Here's what happens when you point BIOS at it.
Step 1: See the structure
First, we need the shape of the lock.
BIOS pulls the target's record (for GLP-1R, UniProt P43220: a 463-residue Class B1 GPCR with 62 deposited PDB structures) and predicts its full 3D fold with AlphaFold, mapping amino-acid sequence to folded structure. We don't hand you that structure as if it were settled truth. Two per-prediction confidence signals are rendered right alongside it:
- pLDDT (predicted local distance difference test): a per-residue confidence score from 0 to 100, painted onto the backbone. Above ~90 (dark blue) the local geometry is essentially trustworthy; the yellow-to-red stretches (below ~70) are where the model is hedging, usually flexible loops you don't want to anchor a binder to.
- PAE (predicted aligned error): the expected positional error, in ångströms, for every residue pair. It's what tells you whether two domains are confidently placed relative to each other, not just well-folded locally. BIOS renders the full residue-by-residue heatmap.
A prediction is a hypothesis, not a fact, and these two scores are how we keep that honest. They decide how much weight a downstream keyhole earns.
Step 2: Find the keyholes
Now we look for the places worth aiming at: the binding sites, or hotspots.
We don't trust a single source for this. BIOS fuses four independent evidence tracks, each rendered as its own annotation lane over the sequence:
- P2Rank: a machine-learning model that predicts and ranks ligand-binding pockets directly from the 3D structure, no homology required;
- Co-crystal data: residues lifted from experimentally solved complexes (for GLP-1R, structures like PDB 7KI0), where the lab has already caught a ligand sitting in the keyhole;
- Literature & mutagenesis: residues that published alanine scans and binding assays proved actually matter: mutate them and binding drops;
- Known GPCR pocket & contact residues: the conserved Class B1 architecture (such as the ECL1 cluster) drawn from decades of receptor pharmacology.
Each candidate residue is scored by how strongly the tracks agree on it. On this run, 46 of 81 scored residues clear the high-confidence bar (score ≥ 2.0), written like R310, E128, D215: the letter is the amino acid (R = arginine, E = glutamic acid, D = aspartic acid), the number is its position along the 463-residue chain. Those high-confidence hits are pre-selected, then BIOS layers P2Rank's predicted binding pocket on top, so the keyhole set passed to the design stage is broader than the ≥ 2.0 tier alone. Here the Generate Binders (123) button hands 123 residues forward.
For a protein this well-characterized, the evidence converges: when P2Rank, a co-crystal, and a mutagenesis hit all land on the same residue, a key built for it starts with strong odds.
Step 3: Make the keys
This is the 90%.
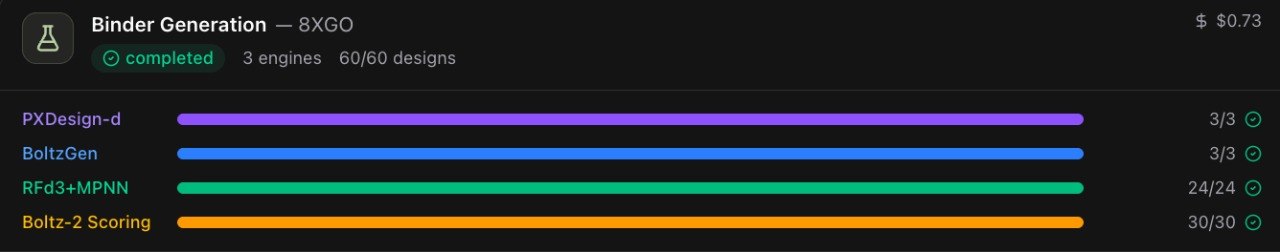
When you click Generate Binders, BIOS doesn't ask one model for an answer. It runs three architecturally distinct generators in parallel on every campaign, each inverting the problem AlphaFold solves, going from the keyhole's geometry to a sequence that fills it:
- RFd3 + MPNN: RFdiffusion3 diffuses a backbone shaped to the pocket, then ProteinMPNN solves the inverse-folding step, assigning the amino-acid sequence most likely to fold into that backbone;
- BoltzGen: a generative model that designs the binder against the target in one shot;
- PXDesign-d: a separate design model run with an independent prior.
They're three locksmiths trained at different benches: their failure modes don't correlate, so a pocket that defeats one is often solved by another, and we never know in advance which. That non-correlation is the whole point. Bet on a single generator and you inherit a single model's blind spots; run three and pool the output and design diversity stays high at the source. A full campaign produces 100 candidate binders across the three engines (short peptides, typically 30–40 residues), streamed into the scoring stage and ranked live by iPTM as they land.

Step 4: Score and filter
We now have 100 keys. Most of them are no good. The art is throwing the bad ones away, fast, and for the right reasons.
A key principle runs through everything here: the models that judge a design are never the models that made it. Generate and score with the same family and they agree with themselves, and you get fooled. Mixing independent, state-of-the-art predictors keeps that self-confirmation bias low.
The first cut runs live, during generation: a cheap triage gate of pLDDT ≥ 70%, iPTM ≥ 50%, iPAE < 10 that drops obviously misfolded or non-binding designs before they cost anything. What survives goes through three deeper layers:
- Structural confidence: does this key actually fit the lock? Every design is co-folded against the target by Boltz-2, which returns iPTM, ipSAE, pLDDT, interface RMSD, and a predicted binding free energy. Each is then re-folded independently by ESMFold2-Fast across three diffusion seeds. When two structurally independent predictors converge on the same binding pose, confidence compounds; when they disagree, we flag it, catching the false positives that single-model gating sails right past.
- Selectivity: does this key open only the right lock? A key that opens every door is useless and dangerous. Each surviving design is screened against 8 paralog receptors (the target's closest structural relatives), pulled automatically with mmseqs2 (by sequence) and Foldseek (by structure) from a 398-GPCR database. A Δ-iPTM ≥ 0.2 gate rejects anything that binds a neighbor nearly as tightly as the target. For GPCR peptide drugs, cross-reactivity is the dominant failure mode, so we screen it out before anything reaches a bench.
- Physics: would it hold together in the body? Beyond the learned models, physics-based contact analysis (PRODIGY, FreeSASA) estimates binding affinity, buried surface area, and interface energy (how hard the key actually grips), with molecular-dynamics stability checks available for a closer look.
The result is a short, ipSAE-ranked list of the strongest candidates: the keys that the generators, two independent structure predictors, and the physics all agree should fit.

On metrics: validated, not invented
One more thing about that scoring stack, because it's where a lot of platforms quietly cheat.
There are dozens of interface-confidence metrics (iPTM, iPAE, ipSAE, pLDDT, interface RMSD), and it's easy to pick the threshold on whichever one flatters your results. We don't.
The fast in-loop filter leans on iPTM and iPAE because they're cheap to compute at generation time. But the metric we rank on is ipSAE, the score the Overath et al. meta-analysis of 3,766 experimentally tested binders identified as the single strongest predictor of real wet-lab binding success, with 1.4× the precision of iPAE. Every cutoff is calibrated from those real binding distributions, not copied out of a paper's defaults.
So when BIOS tells you a design is promising, that confidence traces back to thousands of physical experiments, not to a number we found convenient.
The same pipeline, a different lock
Here it is end to end on a completely different one: KISS1R, the kisspeptin receptor (PDB 8XGO), a Class A peptide GPCR rather than GLP-1R's Class B1. A single campaign fired all three generators, co-folded and scored every design through Boltz-2, and surfaced a ranked shortlist — start to finish for $0.73 (a smaller, quicker run than the 100-design GLP-1R campaign above).
The scored designs land in a live grid, ranked by iPTM and color-coded by ipSAE — green for the strong binders, amber and red for the ones the gate is about to drop:
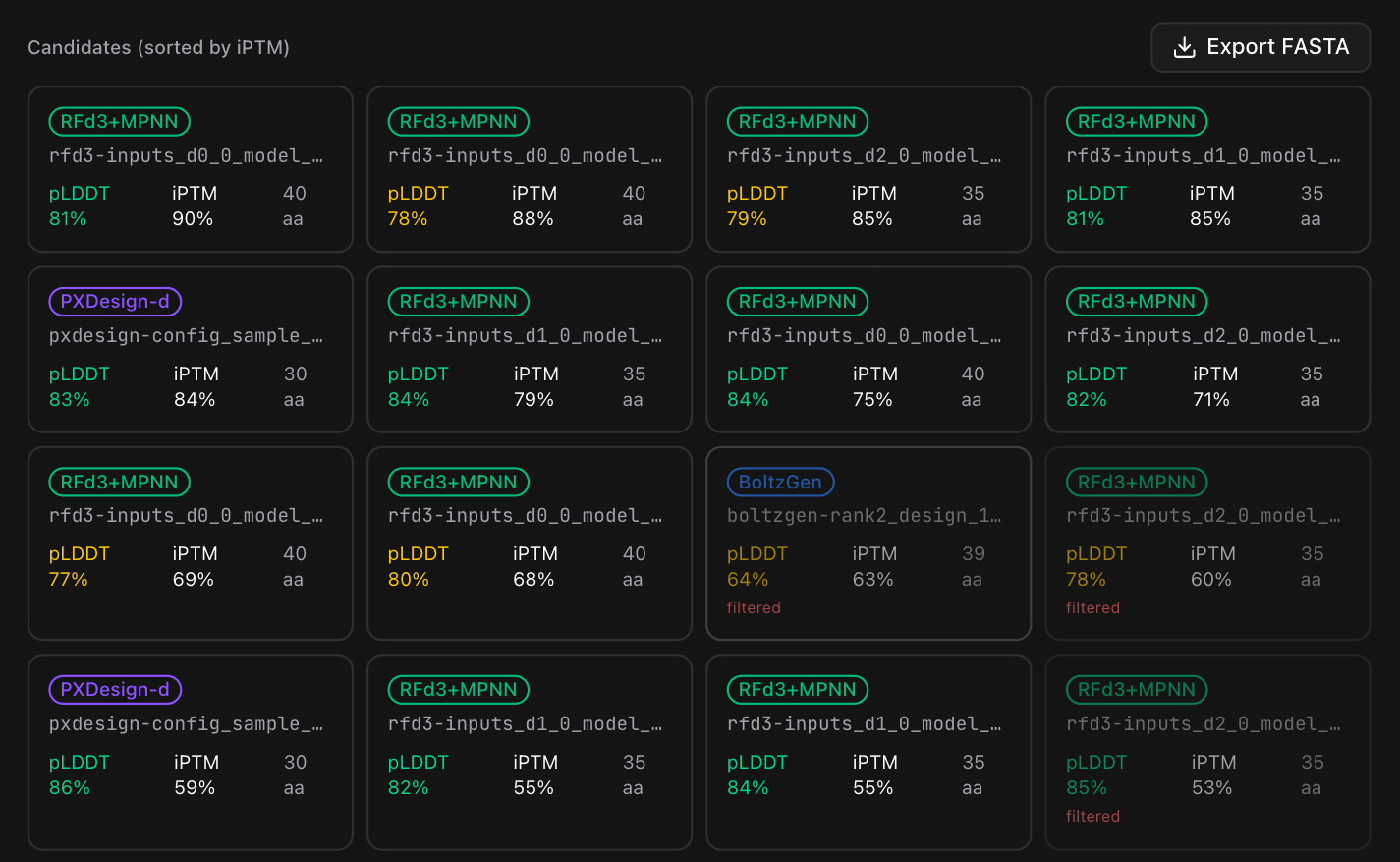
Open any one and you get its full scorecard — the rendered fold, every interface metric, the sequence, and a verdict on whether it advances:
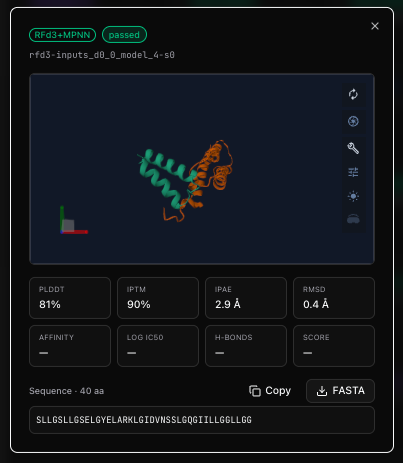
The robot: closing the loop
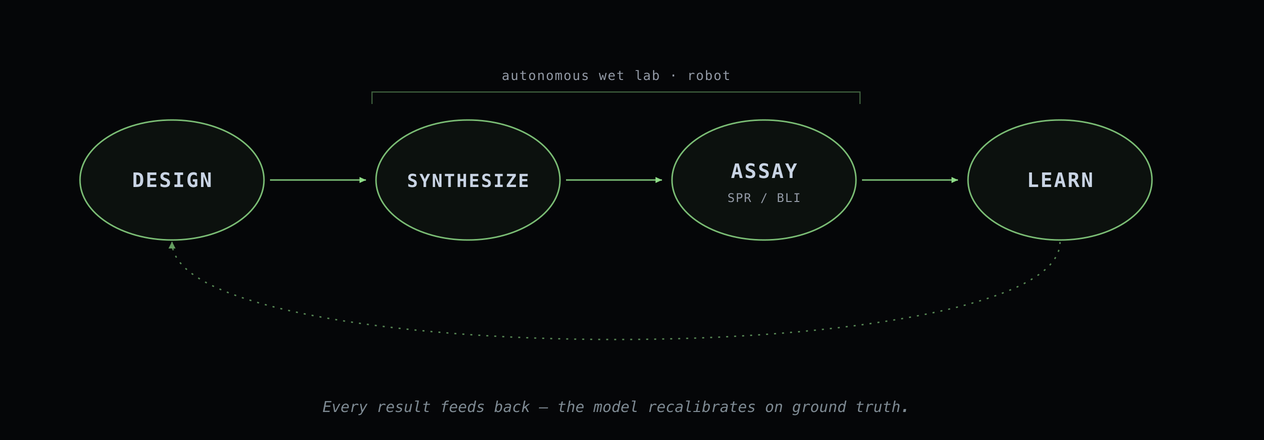
A predicted binder is still just a hypothesis. So we're building the hardware to test it.
Our autonomous wet-lab system takes BIOS's top-ranked sequences and produces purified peptide vials (solid-phase synthesis, HPLC purification, mass-spec QC), running unattended. A robotic SPR/BLI station then measures how tightly each candidate actually binds, against both the real target and a panel of those paralog counter-targets.
And then the important part: every result feeds back.
Hit or miss, the data returns to the model. ipSAE thresholds recalibrate. Composition filters tighten. The system learns which features predict real binding for each class of target. Like telling an image generator "more like this one," except the feedback is ground truth from a robot, not a vibe.
Design → Build → Test → Learn → Repeat.
When that loop runs in days instead of years, the rate of discovery stops being limited by human hands. It starts being limited only by how fast we can think.
The vision: medicine designed for one
Here's where this goes.
GPCRs (the family GLP-1R belongs to) carry thousands of missense variants across the human population. Tiny spelling changes in the genetic code that reshape a drug's binding pocket. They're a big part of why the same drug works beautifully for one person and does nothing for another.
Now imagine a platform that designs binders per variant. You feed in a patient-derived target. In under an hour, you get candidates tailored to that exact pocket. They're synthesized on-site, validated robotically, and iterated, until one is right.
That's not a research tool. That's a new kind of medicine.
The compute cost is already $7 per patient-specific campaign. The synthesis-and-assay turnaround is what our hardware closes. BIOS is the design engine of that future.
We're also building BIOS to be open to anyone who's curious: researchers, computational biologists, and people who just want to see how a medicine gets made. Designing therapeutics has been locked behind the walls of a handful of institutions. We'd like to hand the keys out.
Design. Synthesize. Validate. Learn. Repeat.
Follow the work on X — @BioAIDevs · @joweniger · @GeeveGeorge
bios.dev · Berlin / Bengaluru · June 2026